Diagnosis of Dermatomyositis & EULAR classification
Early diagnosis and treatment of dermatomyositis is essential for optimal prognosis.1-3 Delaying treatment of dermatomyositis can lead to irreversible muscle damage and persistent muscle weakness.4
Diagnosis of dermatomyositis is challenging because of the heterogeneity between myositis subtypes3, and overlap with symptoms from a number of other diseases.1
There is a lack of clear diagnostic criteria for idiopathic inflammatory myopathies.3
Additionally, multisystem involvement can complicate the diagnostic process, with wide variation in how the different subtypes manifest.3
Diagnosis of idiopathic inflammatory myopathies includes assessment of the clinical history, disease progression, and pattern of muscle involvement and/or cutaneous features.5
Except for some cases of dermatomyositis that have a typical presentation, diagnosis is usually not straightforward and requires a combination of tests.4
A number of diagnostic tests are available for patients where dermatomyositis is suspected, including serum muscle enzymes (e.g. creatine kinase), myositis antibodies, electrodiagnostic studies, muscle biopsy and muscle MRI.6
Muscle biopsy is the gold standard for diagnosis of dermatomyositis.2
Several criteria have been developed to diagnose dermatomyositis, including the Bohan and Peter Criteria2,7,8; however, these have not been validated.9
Although the Bohan and Peter Criteria demonstrate a high degree of accuracy, they have limitations.3 For example, they do not include the use of myositis-specific autoantibodies to identify the specific idiopathic inflammatory myopathy subtype3, which are important for diagnosis.
More recently, EULAR/ACR classification criteria were developed for idiopathic inflammatory myopathies and these criteria have been partially validated and been shown to generally perform better than existing criteria.9

There is a lack of clear diagnostic criteria for idiopathic inflammatory myopathies.3
EULAR classification tree for subgroups of idiopathic inflammatory myopathies (IIMs)3
The European League Against Rheumatism (EULAR) classification tree for subgroups of idiopathic inflammatory myopathies is a tool that helps classify and diagnose idiopathic inflammatory myopathies, which are a group of rare autoimmune disorders that cause muscle inflammation and weakness. The classification tree is structured as follows:
 including dermatomyositis")
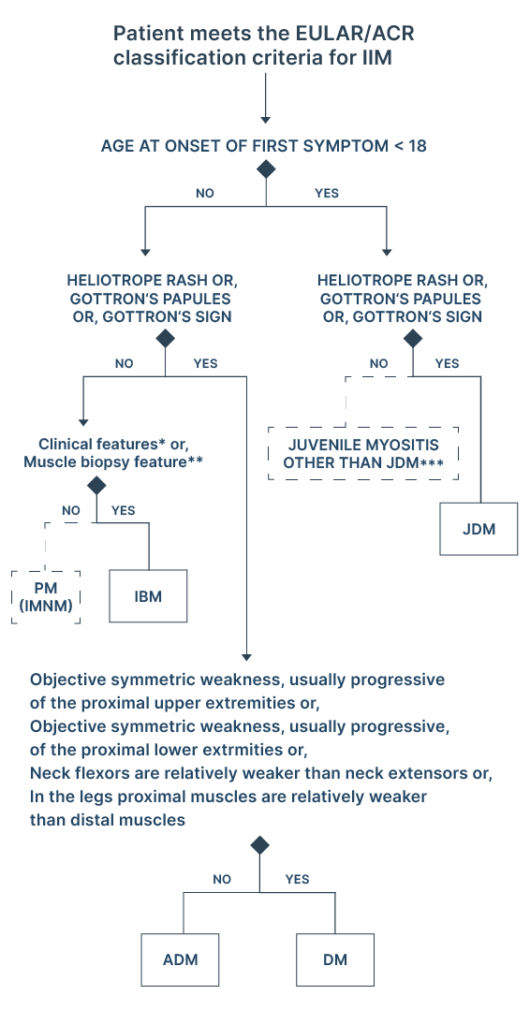
Adapted from EULAR publication11
Classification tree for subgroups of idiopathic inflammatory myopathies (IIMs). A patient should first be classified as having IIM using the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) classification criteria.
The patient can then be subclassified using the classification tree. The mixed (dotted outlined box) subgroup of patients with PM includes patients with IMNM. For IBM diagnosis, one of the following,
*Finger flexor weakness and response to treatment: not improved or
**Muscle biopsy: rimmed vacuoles, is required for diagnosis.
***Juvenile myositis other than JDM was developed based on expert opinion and extrapolation from adults. IMNM and hypomyopathic DM were too few to allow subclassification.
ADM, amyopathic dermatomyositis
DM, dermatomyositis
IBM, inclusion body myositis
IMNM, immune-mediated necrotising myopathy
JDM, juvenile dermatomyositis
PM, polymyositis
Tools to aid dermatomyositis diagnosis
There are several tools that can be used to aid in the diagnosis of dermatomyositis, a type of idiopathic inflammatory myopathy (IIM). These tools include:
Serum muscle enzymes
In dermatomyositis, serum creatine kinase (CK) levels are often elevated due to muscle membrane damage and muscle necrosis.6
However, in some patients CK levels are normal, and CK levels are not useful in monitoring disease progression or activity.
Other enzymes released from damaged skeletal muscle can also be increased in patients with dermatomyositis:
- Aldolase
- Lactate dehydrogenase
- Alanine aminotransferase
- Aspartate aminotransferase
Electrodiagnostic studies
Electromyography (EMG) changes are non-specific and not useful for differentiating between myositis subtypes.4 EMG is useful to:
- Confirm a myopathic process6
- Rule out neurogenic conditions associated with proximal muscle weakness6
- Distinguish between acute changes in myositis and steroid myopathy weakness4
In patients with myositis, findings from nerve conduction studies are typically normal.6 However, when myositis-associated weakness is severe and diffuse, low-amplitude motor nerve responses can be seen.
Muscle biopsy
- Muscle biopsy is the gold standard for diagnosing dermatomyositis2
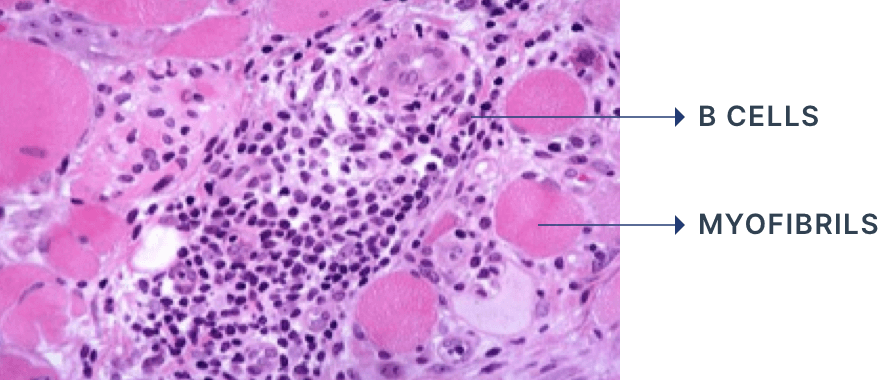
- Perifascicular muscle fibre atrophy is a specific, histopathological feature of dermatomyositis6
- Inflammatory infiltrates are predominantly located in the perimysium and perivascular regions and include CD4+ T cells2,6, B cells2 and plasmacytoid dendritic cells6
- A reduced number of capillaries can be observed6
- Muscle fibres show evidence of necrosis and phagocytosis5
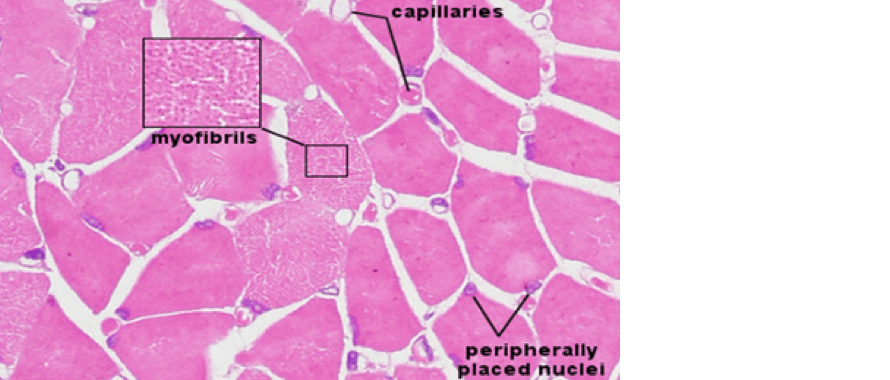
Histological section of healthy skeletal muscle
Histological section of dermatomyositis skeletal muscle
Myositis autoantibodies
- Five known dermatomyositis-specific autoantibodies (Mi-2,TIF-1γ, NXP-2, MDA-5 and SAE) are associated with distinct clinical phenotypes and may be useful prognostic markers6
- Identification of myositis-specific autoantibodies can:
- Aid earlier diagnosis in myositis patients
- Guide clinical management based on autoantibody subtypes with the associated risks of malignancy, risks of extramuscular/organ involvement, and response to specific types of immunotherapy
Overview of dermatomyositis autoantibodies and their respective organ involvement6,10
| Antibody | Organ involvement | Features |
| Anti-Mi2 | Good prognosis, favourable response to steroids, relatively low malignancy risk | |
| Anti-TIF1γ | Highly associated with malignancy in adults and severe skin manifestations | |
| Anti-NXP2 | Subcutaneous calcifications in juvenile and adult dermatomyositis, severe dermatomyositis, increased risk of malignancy in adults | |
| Anti-MDA5 | Minimal muscle involvement, rapidly progressive interstitial lung disease and poor prognosis, severe skin lesions | |
| Anti-SAE | May present as amyopathic initially, severe rash, high incidence of dysphagia |
M = muscle / S = skin / L = lung / C = cancer
Muscle magnetic resonance imaging (MRI)
Non-invasive tool to:
- Assess muscle inflammation (manifested by e.g. oedema, atrophy or fatty replacement)6
- Assess the severity of muscle involvement, even at the subclinical level4
- Help localise affected muscles to provide guidance on where to perform a biopsy6
- Monitor a patient’s response to immunotherapy6
- Monitor for acute damage and current inflammation4

One of the main challenges in diagnosing dermatomyositis is that the signs and symptoms of the condition can vary widely from person to person and may be similar to those of other autoimmune disorders or muscle diseases. This can make it difficult to accurately diagnose the condition based on symptoms alone.3
In addition, dermatomyositis can involve multiple systems in the body, such as the skin, muscles, and lungs, which can further complicate the diagnosis.3,4
References
- Briani, C., Doria, A., Sarzi-Puttini, P., & Dalakas, M. C. (2006). Update on idiopathic inflammatory myopathies. Autoimmunity, 39(3), 161–170. https://doi.org/10.1080/08916930600622132
- Malik, A., Hayat, G., Kalia, J. S., & Guzman, M. A. (2016). Idiopathic Inflammatory Myopathies: Clinical Approach and Management. Frontiers in Neurology, 7. https://doi.org/10.3389/fneur.2016.00064
- Oldroyd, A. and Chinoy, H. (2018). Recent developments in classification criteria and diagnosis guidelines for idiopathic inflammatory myopathies. Current Opinion in Rheumatology, 30(6), pp.606–613. doi:https://doi.org/10.1097/bor.0000000000000549.
- Schmidt, J. (2018). Current Classification and Management of Inflammatory Myopathies. Journal of Neuromuscular Diseases, 5(2), 109–129. https://doi.org/10.3233/jnd-180308
- Dalakas, M.C. (2015). Inflammatory Muscle Diseases. New England Journal of Medicine, 372(18), pp.1734–1747. doi:https://doi.org/10.1056/nejmra1402225.
- Goyal, N. A. (2019). Immune-Mediated Myopathies. CONTINUUM: Lifelong Learning in Neurology, 25(6), 1564–1585. https://doi.org/10.1212/con.0000000000000789
- Bohan, A., & Peter, J. B. (1975). Polymyositis and dermatomyositis (first of two parts). The New England Journal of Medicine, 292(7), 344–347. https://doi.org/10.1056/NEJM197502132920706
- Bohan, A., & Peter, J. B. (1975). Polymyositis and Dermatomyositis. New England Journal of Medicine, 292(8), 403–407. https://doi.org/10.1056/nejm197502202920807
- Lundberg, I. E., Tjärnlund, A., Bottai, M., Werth, V. P., Pilkington, C., Visser, M. de, Alfredsson, L., Amato, A. A., Barohn, R. J., Liang, M. H., Singh, J. A., Aggarwal, R., Arnardottir, S., Chinoy, H., Cooper, R. G., Dankó, K., Dimachkie, M. M., Feldman, B. M., Torre, I. G.-D. L., & Gordon, P. (2017). 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Annals of the Rheumatic Diseases, 76(12), 1955–1964. https://doi.org/10.1136/annrheumdis-2017-211468
- Ghirardello, A., Borella, E., Beggio, M., Franceschini, F., Fredi, M., & Doria, A. (2014). Myositis autoantibodies and clinical phenotypes. Autoimmunity Highlights, 5(3), 69–75. https://doi.org/10.1007/s13317-014-0060-4
- Bottai, M., Tjärnlund, A., Santoni, G., Werth, V.P., Pilkington, C., de Visser, M., Alfredsson, L., Amato, A.A., Barohn, R.J., Liang, M.H., Singh, J.A., Aggarwal, R., Arnardottir, S., Chinoy, H., Cooper, R.G., Danko, K., Dimachkie, M.M., Feldman, B.M., García-De La Torre, I. and Gordon, P. (2017). EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups: a methodology report. RMD Open, [online] 3(2), p.e000507. doi:https://doi.org/10.1136/rmdopen-2017-000507.
Science Hub
Sign up and connect to Science Hub to learn more about our recent ProDERM study and have access to symposia presentations, publications, posters and lectures.